Построение филогенетических деревьев

Самым простым подходом к построению филогенетических деревьев является применение алгоритмов иерархической кластеризации, используя в качестве меры расстояния результат выравнивания последовательностей. Так, применение алгоритма UPGMA (см. подпараграф 2.8.2) в филогении позволяет получать укорененные деревья, у которых длина ветвей соответствует времени «молекулярных часов», прошедшему… Читать ещё >
Построение филогенетических деревьев (реферат, курсовая, диплом, контрольная)
Алгоритмы, оперирующие матрицей расстояний.
Самым простым подходом к построению филогенетических деревьев является применение алгоритмов иерархической кластеризации, используя в качестве меры расстояния результат выравнивания последовательностей. Так, применение алгоритма UPGMA (см. подпараграф 2.8.2) в филогении позволяет получать укорененные деревья, у которых длина ветвей соответствует времени «молекулярных часов»[1], прошедшему от соответствующего эволюционного события.
Другим восходящим иерархическим алгоритмом, о котором стоит рассказать, является алгоритм присоединения соседей {Neighbor joining, NJ). Технологически алгоритм NJ отличается UPGMA-подходом к обработке данных матрицы расстояний, что позволяет избежать неявного предположения о постоянстве скорости молекулярной эволюции. Еще одним отличием алгоритма NJ является то, что он строит неукорененные деревья. Алгоритм состоит из следующих шагов:
1) на основании матрицы расстояний рассчитывается Q-матрица (см. ниже);
- 2) ищется пара узлов (г, у), для которых значение Q матрицы (Q (i, j)) минимально, и эти два узла соединяются с вновь созданным узлом (являющимся их LCA);
- 3) расстояния от выделенных узлов (i, j) до нового (к) рассчитываются по формулам

4) выделенные узлы (i, j) в матрице расстояний заменяются на новый (k), и расстояния до остальных узлов (и) заново рассчитываются по следующей формуле:
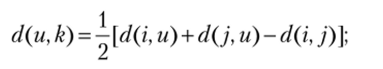
5) если в матрице расстояний остались всего два узла, то алгоритм завершен, иначе переходим к пункту 1.
Расчет Q-матрицы производится по следующей формуле:
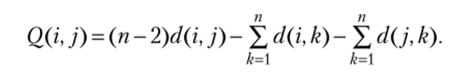
Алгоритмы, анализирующие характер замен. Другим подходом к генерации филогенетических деревьев явился отказ от «слепого» сравнения данных о расстоянии между последовательностями, полученного в результате выравнивания последовательностей и фактически усредняющих информацию по всем сайтам последовательностей, в пользу анализа характера изменения отдельных позиций.
Алгоритмы минимизации количества замен (maximum parsimony — максимальная экономия) нацелены на группировку последовательностей по расстоянию «редактирования», т. е. минимизируют количество замен остатков в ветвях получающегося дерева.
Работа алгоритмов этой группы основана на идентификации так называемых информативных сайтов — позициях в анализируемых последовательностях, для которых возможно построение эволюционных деревьев, неэквивалентных друг другу, с учетом указанного выше принципа. Поясним это на примере. Так, для множественного выравнивания, изображенного на рис. 3.48:
- • позиция 1 не является информативной, так как там нет замен, требующих объяснения;
- • позиция 2 также не является информативной, но уже по причине того, что все три возможных эволюционных дерева, объясняющих данный сайт, включают одну замену;
- • позиции 3 и 4 не являются информативными, так как все возможные эволюционные деревья для этих сайтов подразумевают две и три замены соответственно;
• только сайт 5 является информативным, поскольку одно из эволюционных деревьев подразумевает одну замену, а два других — по две.
Поскольку поиск эволюционного дерева, отвечающего принципу максимальной экономии, является NP-сложной проблемой, большинство алгоритмов данной группы — разного рода эвристики.
Позиция выравнивания.

Рис. 3.48. Оценка информативности позиций выравнивания для метода.
максимальной экономии Алгоритмы, реализующие метод наибольшего правдоподобия в качестве критерия, подлежащего оптимизации, используют соответствие генерируемого дерева определенной модели эволюции (см. далее), позволяющей оценить вероятности одиночных замен. Данный метод оценивает альтернативные гипотезы (эволюционные деревья и параметры эволюционной модели), выбирая наиболее правдоподобный вариант, т. е. дерево, лучшим образом объясняющее исходное выравнивание с позиций используемой эволюционной модели.
Рассмотрим гипотетическое дерево на рис. 3.49. Допустим, у нас определена некоторая функция Рау (Ь)у задающая вероятность замены остатка а на остаток b за временной интервал t. Тогда функция правдоподобия могла бы выглядеть следующим образом: 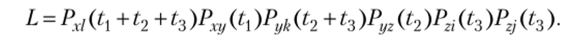
Однако ввиду того что в реальной ситуации филогенетического анализа нам известны только остатки i, j, k и /, то полная функция правдоподобия должна включать суммирование вероятностей для всех вариантов распределения мономеров по внутренним узлам.

Рис. 3.49. Иллюстрация к методу максимального правдоподобия.
Учитывая множество возможных топологий, а также необходимость включать в расчет длины ветвей, поиск решения в данных алгоритмах производится с применением стратегий глобальной оптимизации, ищущих параметры модели, отвечающие максимуму значения L.
Молекулярные модели эволюции. Поскольку большинство современных методов проведения молекулярно-филогенетических построений в явной форме пытаются оперировать возможными сценариями эволюционной истории, рассмотрим ниже варианты решения данного вопроса.
Первые эволюционные модели замен в полимерах оперировали отдельными остатками. При этом исторически при анализе последовательностей ДНК был в ходу теоретический подход:
- • модель Джукса — Кантора, подразумевавшая одинаковые (равные) частоты замен нуклеотидов;
- • двухфакторная модель Кимуры, полагавшая разные частоты для транзиций и трансверсий;
- • более поздние попытки учитывать колонную структуру кодирования белков.
Что касается анализа белковых последовательностей, то там сразу возобладал практический подход — с классическим примером моделью Дейхофф, с которой мы уже сталкивались, когда рассматривали способ построения матриц замещения РАМ.
Упомянутые выше ранние разработки предполагали независимые частоты замен мономеров, что является довольно грубым приближением с многих точек зрения:
- • индивидуальные частоты мутаций в ДНК зависят от типичной для данного фрагмента плотности упаковки молекулы;
- • замены, с которыми имеет дело молекулярная филогения, «зафиксировались» в ходе эволюции, а значит, прошли через давление отбора, действующего на множестве уровней.
Данные наблюдения привели к зарождению в начале 1990;х гг. ряда новых моделей, сначала учитывавших только простые эффекты, такие как разные частоты мутаций в кодирующих и некодирующих участках ДНК. Развитие этих моделей привело к появлению работ с применением скрытых моделей Маркова для анализа отдельных сайтов, а также по включению данных структурной биологии (структурное окружение/вторичная структура РНК) в учитываемые факторы.
- [1] Гипотеза «молекулярных часов» предполагает постоянную скорость молекулярнойэволюции, что является очень грубым приближением реальных эволюционных механизмов.